COMPARING MICROBIOLOGICAL TEST METHODS – PART 1

Culture Versus Non-Culture Test Methods
History
There is a false impression among microbiologists and non-microbiologists alike that because culture testing has been around since the mid-19th century, it is a reference method (I’m come back to the reference method concept in a bit). The first quantitative culture-based method – the heterotrophic plate count (HPC) first appeared in Standard Methods for the Examination of Water and Wastewater Standard Methods, 11th Edition, 1960. Since then, thousands of variations of the HPC method have been developed. They differ in the nutrients used (1000s of different recipes), growth conditions under which inoculated Petri plates are incubated (100s of temperature, relative humidity, and gas combinations), and how the specimen is introduced to the medium (pour plate, spread plate, and streak plate methods). Because of the variety of plate count protocols, ASTM offers a practice rather than a test method – D5465 Standard Practices for Determining Microbial Colony Counts from Waters Analyzed by Plating Methods.
Alternative Tools for Measuring Microbial Bioburdens
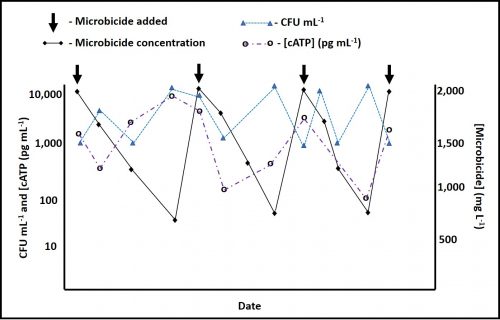
Between 2016 and 2018 I wrote a series of articles in which I explained common types of microbiological test methods (see What’s New from Early and Late December 2016, February, Early and Late July, and August 2017, January and February 2018). As I wrote in 2016, each method contributes to our understanding of microbial contamination. Although each quantifies a different property of microbial bioburden (i.e., the number of microbes present or the concentration of a chemical that is tends to be proportional to the number of microbes present), the data generated by different methods generally agree. As new methods are used, analysts invariably want to know how the results compare against those obtained by culture testing. ASTM E1326 Standard Guide for Evaluating Non-culture Microbiological Tests reviews consideration that should be taken into account when either evaluating the reliability of a new test method or choosing among available methods.
Reference Test Method
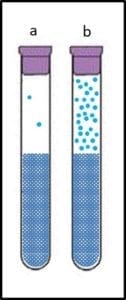
A reference test method is one that is known to provide the most accurate and precise indication of the parameter being tested. Accuracy is the degree to which a measurement or test result agrees with the true or accepted value (for example, an atomic clock – accurate to 10−9 seconds per day – is more accurate than a spring-mechanism wristwatch – the best of which are accurate to 1 second per day). Precision is the degree to which repeated measurements agree. Figure 1 illustrates these two concepts. In Figure 1a, the results (dots on the target) are both accurate – clustered around the bullseye – and precise – close together. The dots in Figure 1b are precise, but not accurate – the cluster is distant from the bullseye. The dots in Figure 1c are accurate – they are near the bullseye, but not particularly precise. To assess a method’s accuracy, you must first have a reference standard – a substance with known properties (for time, the internationally recognized reference standard is the second – 9 192 631 770 cycles of radiation corresponding to the transition between two energy levels of the ground state of the cesium-133 atom at 0 °K). There is no reference microbiological test method because:
- Under a given set of conditions, different microbes will behave differently. Test results will be affected by the types of microbes present.
- A given microbe will behave differently as test conditions vary.
- There is no consensus standard, reference microbe.

A consensus standard is one that has been developed and approved under the auspices of a standards development organization such as ASTM, AOAC, ISO, and others. Consensus standard test methods include precision statements that cite interlaboratory study-based repeatability and reproducibility variation, and bias.
Repeatability is a measure of the variability of results obtained by a single analyst testing replicate specimens from a single sample, using a single apparatus and reagents from single lots. Figure 2 illustrates the repeatability variability for an adenosine triphosphate (ATP) test performed on a metalworking fluid sample. The results are Log10 pg mL-1 where REP is the replicate number and [cATP] is the concentration of cellular ATP per ASTM Test Method E2694.

In comparison, nominal HPC repeatability variation is approximately half an order of magnitude (0.5 Log10 CFU mL-1, where CFU is colony forming units).
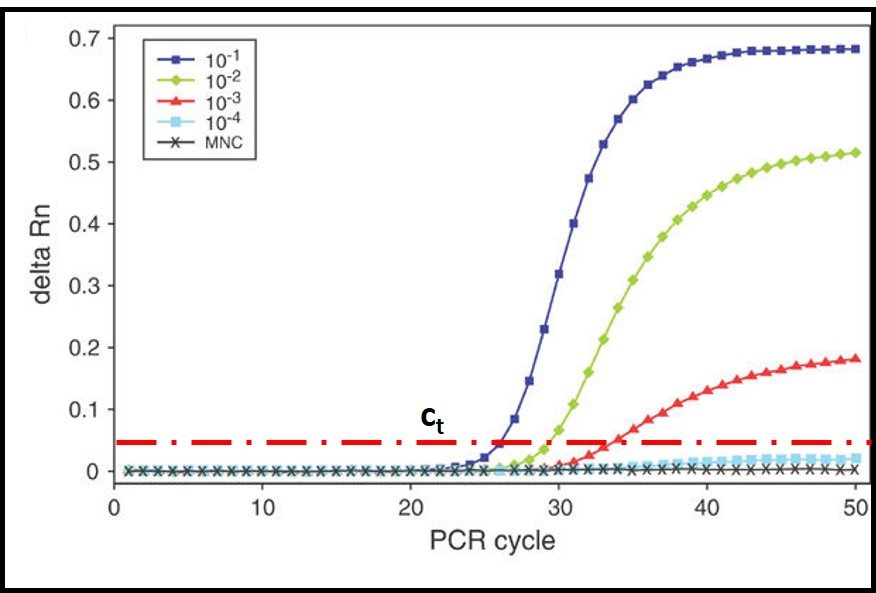
Reproducibility is a measure the variability among multiple analysts running replicate tests on specimens from a single sample, using different sets of apparatus, and reagents. For stable and homogeneous parameters (for example specific gravity) analysts participating in a reproducibility evaluation (interlaboratory study – ILS) are at different facilities. Because microbial contamination is neither homogeneous nor stable, reproducibility testing is commonly performed by analysts at different work-stations located at a single facility. This is called single-site reproducibility testing. Figure 3 illustrates the results of ASTM E2696 reproducibility testing. Ten labs participated in the ILS. The reproducibility standard deviation (sR) was 0.39. Invariably, sR is greater than the repeatability standard deviation (sr).

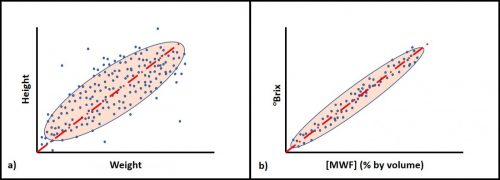
Bias is the difference between a measurement and a parameter’s true value. The cluster of dots in Figure 1b illustrates bias – the distance between the average position of the dots in the cluster and the target’s actual center. Unless there is a reference standard against which a measurement can be compared, only relative bias – the difference between test results obtained by different methods – can be assessed. Figure 4 illustrates bias and relative bias. A water-miscible metalworking fluid (MWF) has been diluted to prepare emulsions with concentrations ([MWF]) of 1, 2, 3, 4 & 5 % v/v. These are the true concentrations ([MWF]T). Each dilution is tested by two methods: refractive index ([MWF]RI) and acid split ([MWF]AS). Each method’s correlation with [MWF]T = 1.0 (0.998 and 0.997 both round to 1.0). However, each has a bias relative to [MWF]T. In this example, [MWF]RI tends to underestimate [MWF] by 16 % and [MWF]AS tends to over-estimate [MWF] by 20 %. The relative bias between the two methods is 36 % – at any [MWF]T, [MWF]AS = 1.20 [MWF]T and 1.36[MWF]RI. Once bias has been determined, it can be used to correct observed values to either true or reference method values.

As illustrated in figure 5, when two methods measure the same parameter, r is normally expected to be ≈1.0. Bias is only meaningful between two methods used to measure the same parameter (i.e., characteristic or property).

The relationship I’ve used [MWF] test methods to illustrate in this What’s New article is similar to what one would expect when comparing two different culture test methods – for example ASTM D6974 Standard Practice for Enumeration of Viable Bacteria and Fungi in Liquid Fuels—Filtration and Culture Procedures and ASTM D7978 Standard Test Method for Determination of the Viable Aerobic Microbial Content of Fuels and Associated Water—Thixotropic Gel Culture Method. Calibration curves based on dilutions of an original sample with a population density of X (in figure 6, X = 320 CFU mL-1; 2.5Log10 CFU mL-1) are expected to have slopes and r-values ≈1. Because bioburdens can range across ≥5 orders of magnitude (i.e., from <10 CFU mL-1 to >106 CFU mL-1) data are commonly converted from linear to logarithmic (Log10) values. The data in figurer 6 meet our expectations. The trendline’s slope (y) = -0.85 ≈ 1 and r = 1.

In my next post, I’ll discuss the relationship between methods that measure different but related properties.
Summary
There are a growing number of test methods that can be used to assess bioburdens. Many of these methods quantify the concentration of a biomolecule or class of biomolecules (adenosine triphosphate, carbohydrates, nucleic acids, proteins, etc.). In this article, I reviewed the basic concepts of data variability – repeatability and reproducibility – and bias, and the expected relationship between two methods that purport to measure the same property (for example, two methods to determine [MWF]). In Part 2 I’ll discuss the principles of comparing different methods for assessing microbial bioburden.
As always, I invite your comments and questions. You can reach me at fredp@biodterioration-control.com.