WHAT’S IN A NAME – ARE BIOCIDE-FREE METALWORKING FLUIDS LESS TOXIC THAN THOSE FORMULATED WITH BIOCIDES?

One of the more famous quotes from William Shakespeare’s play, Romeo and Juliet.
Language Matters
In this month’s article I’ll address the use of what I call unregistered microbicides.
Over the course of the past several decades, industry and regulators have taken increasingly jaundiced views of chemical substances variously known as antimicrobial pesticides, biocidal substances, biocides, biocidal products, and microbicides. What are these substances? The EU’s Biocidal Products Regulation (BPR – Regulation (EU) No 528/2012 of the European Parliament and of the Council of 22 May 2012) Article 3, 1 (a) offers this definition:
— any substance or mixture, in the form in which it is supplied to the user, consisting of, containing or generating one or more active substances, with the intention of destroying, deterring, rendering harmless, preventing the action of, or otherwise exerting a controlling effect on, any harmful organism by any means other than mere physical or mechanical action,
— any substance or mixture, generated from substances or mixtures which do not themselves fall under the first indent, to be used with the intention of destroying, deterring, rendering harmless, preventing the action of, or otherwise exerting a controlling effect on, any harmful organism by any means other than mere physical or mechanical action.
A treated article that has a primary biocidal function shall be considered a biocidal product.
I’ve highlighted key words in the BPR definition because one response from industry has been to replace products that are registered as microbicides with alternative chemistries that do are not registered. They then promote their finish goods as being “biocide free.”
I pose this question:
Is it legitimate to make a biocide-free claim if a substance is used to control microbial contamination in a formulated product although it does not have an antimicrobial pesticide registration?
Non-biocidal additives in water-miscible metalwork fluids (MWF)
There are three groups of products related to MWF biodeterioration resistance – bioresistant, biostatic, and adjuvant additives.
Bioresistant additives
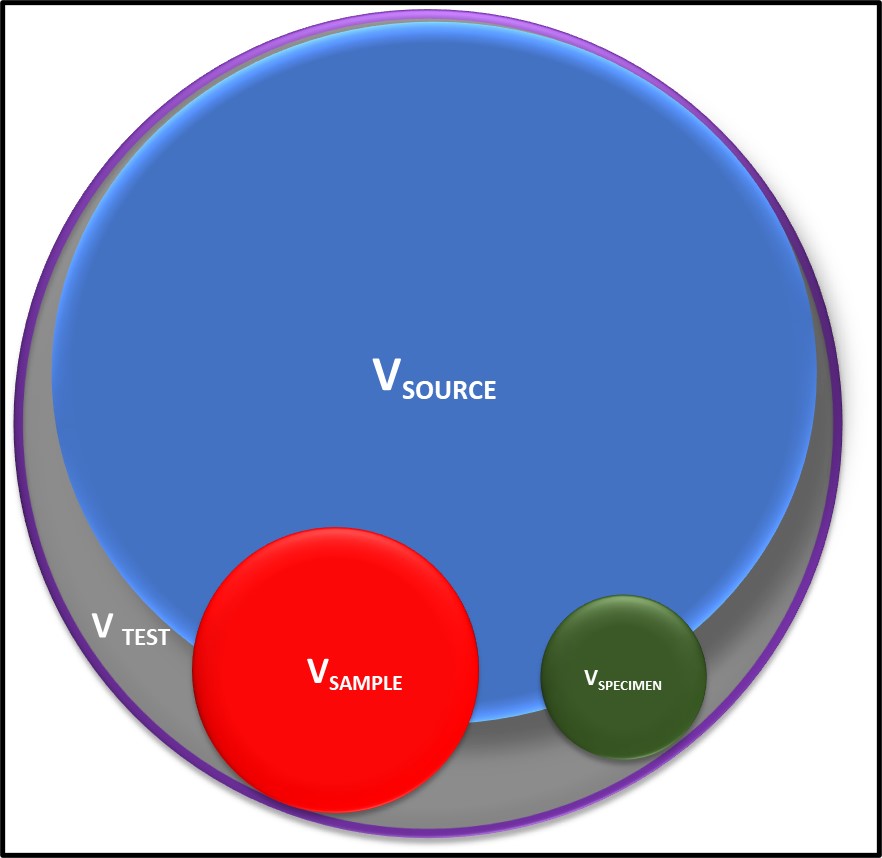
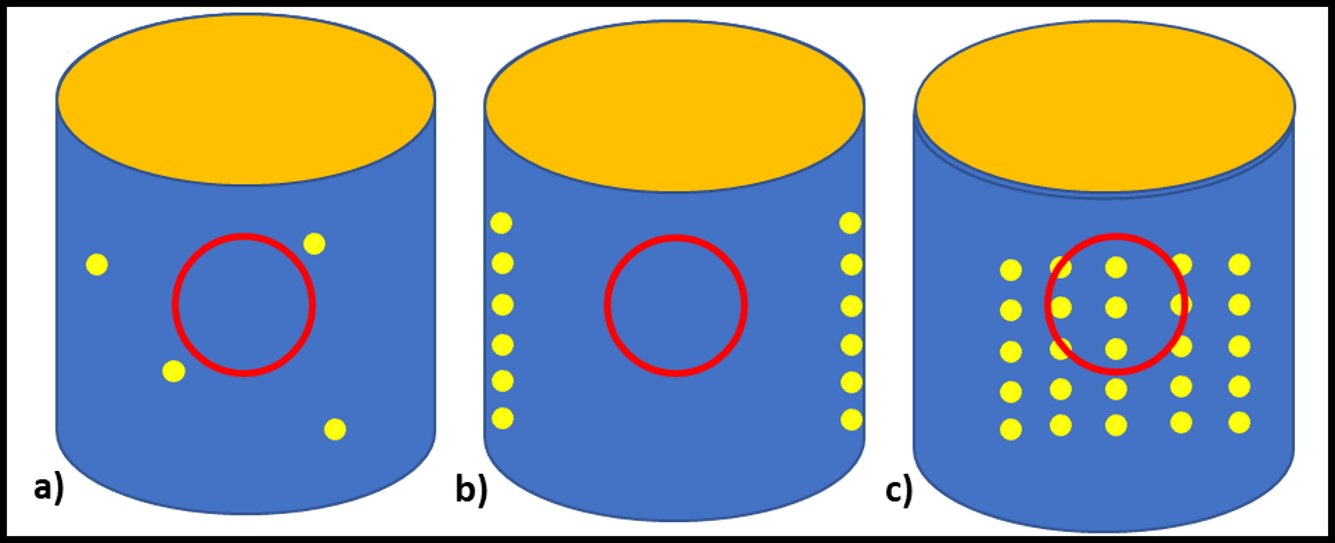
Bioresistant (recalcitrant) additives are chemistries that are difficult for microbes to use as food. As illustrated in Figure 1, their concentration in a fluid is unaffected by the fluid’s bioburden.
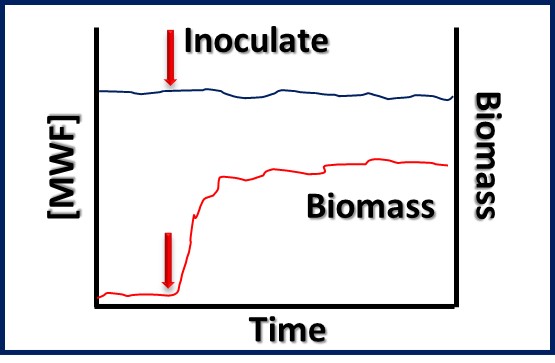
Fig 1. Bioresistant MWF additive – additive concentration is unaffected by microbial load (bioburden).1
Biostatic additives
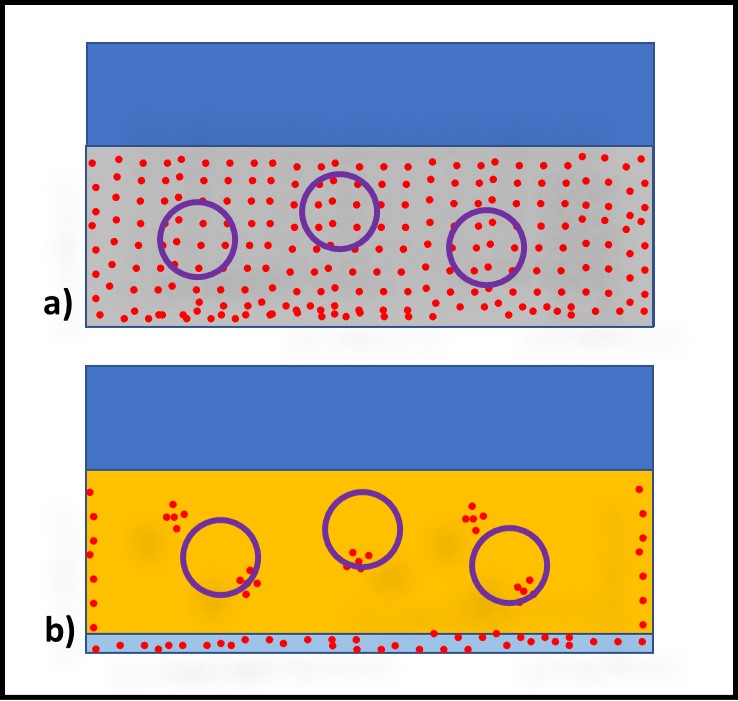
In contrast to bioresistant additives, for which there appears to be no interaction between microbes and the additive, biostatic additives contribute to the MWF formulation’s ability to resist microbial growth. Figure 2a shows that when a biostatic MWF is inoculated with microbes, they do not proliferate (i.e., the biobuden does not increase). However (Figure 2b), if a biostatic additive is added to a heavily contaminated MWF, it has no effect on the biobuden.
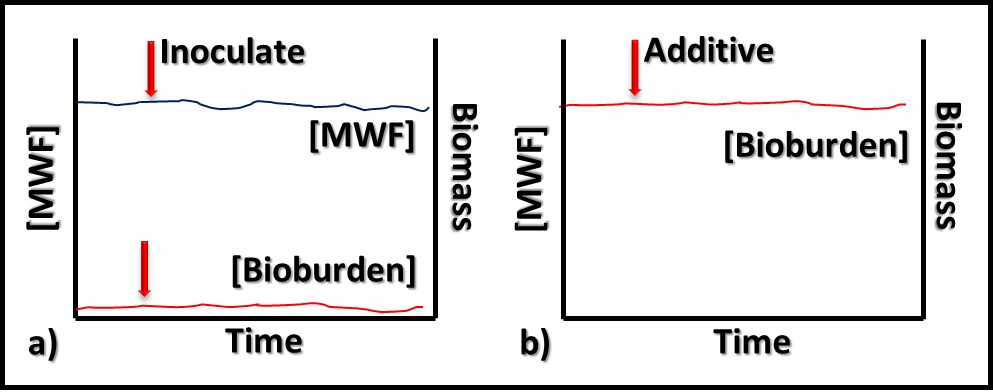
Fig 2. Biostatic MWF additive – a) When microbes are added to biostatic MWF formulation, they do not proliferate; b) when a biostatic additive is added to a heavily contaminated MWF, it has no impact on the bioburden.
Adjuvants
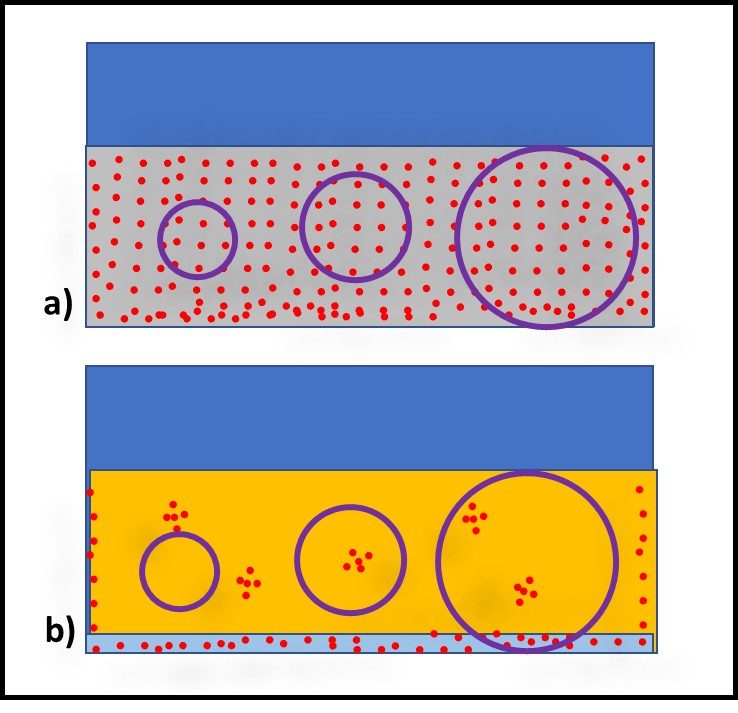
Additives that have no direct impact on microbial contamination in MWF (Figure 3a), but which improve the performance of microbicides are called adjuvants. Figure 3 illustrates this concept. Microbicides can kill off microbes (Figures 3b and 3c, red line), prevent microbes from proliferating (Figure 3d), or do both.
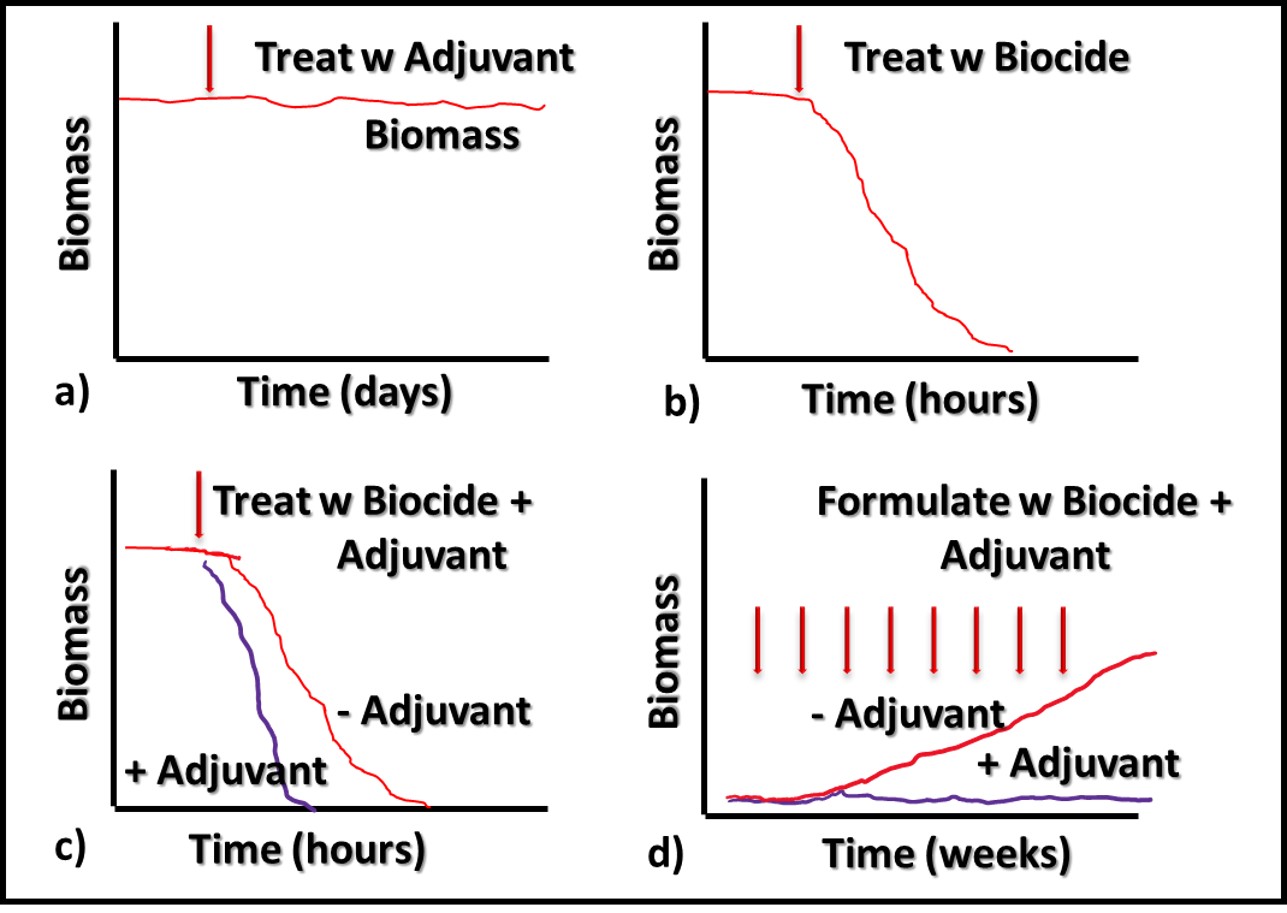
Fig 3. Adjuvant MWF additive impact on biomass – a) adjuvant without microbicide; b) microbicide speed of kill without adjuvant; c) microbicide speed of kill with adjuvant; d) microbial proliferation in MWF formulated with microbicide (red line) and microbicide plus adjuvant (purple line).
Similarly, an adjuvant can increase a microbicide’s speed of kill (Figure 3c, purple line), prolong the duration of its effectiveness against repeated challenges (Figure 3d, purple line) or both. The red arrows in Figure 3d indicate weekly inoculation of the test MWF with a microbial challenge population per ASTM Practice E2275.
Unregistered, microbicidal additives in water-miscible MWF
A key word in BPR’s biocide definition is intention. With this word, BPR’s definition shifts from an objective perspective – if a substance has a controlling effect on microbes, it is a microbicide – to a subjective perspective – only if it was intended for a substance to have a controlling effect on microbes is that substance subject to BPR registration. The U.S. Federal Insecticide, Rodenticide and Pesticide Act (FIFRA) has similar language (Sec. 2 [17 U.S.C. 136 (u)). The challenge is in reaching consensus on the meaning intention regarding the use of MWF functional additives.
Functional additives
In the MWF sector, a functional additive is a chemical substance that provides one or more performance properties to the fished formulation. Typical functional additive performance properties include:
- Corrosion inhibition
- Coupling – additives that provide chemical bonds between dissimilar substances (e.g., base oils and polar molecules)
- Emulsion stabilization
- Foam inhibition
- Lubricity
- Microbicidal activity
- pH control (buffering)
Products used in several of these functional categories also impact microbial contamination. All chemicals sold for use in technical applications Europe must be registered in accordance with Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH – Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006). In the early and mid-1990s, I was hopeful that REACH would the toxicity data required for all industrial chemicals would be similar. This would have closed the cost gap associated with obtaining the toxicity data needed for microbicide registration versus that needed for non-microbicidal substances. However, requiring a full toxicological test package for each of the millions of industrial chemicals was determined to be prohibitively expensive. Additionally, the time and laboratory facilities required to test all industrial chemicals rendered the concept untenable. Consequently, although some toxicological data are required to support product registrations under REACH, substantially more is needed for product registration under BPR. This creates a grey zone.
What is the difference between a registered and an unregistered microbicide?
Per the definition I quoted in the opening paragraph, a registered microbicide is an active substance (ingredient) or formulated product intentionally used to control microbial contamination and approved for such use by the cognizant regulatory agency (e.g., the European Chemical Agency’s – ECHA’s – Biocidal Products Committee, and the U.S. EPA’s Office of Pesticide Programs).
There is no consensus on definition of an unregistered microbicide. Nor is there consensus about the concept of intention. There is no universally agreed upon demarcation between a non-biocidal additive that also affects microbial contamination and one that has some level of non-biocidal activity (e.g., corrosion inhibition) but primarily inhibits microbial growth. To further complicate matters, there are numerous technical grade substances that are substantially more toxic than biocidal products. Moreover, there are registered microbicides that have non-biocidal applications. For example, hexahydro-1,3,5-tris(hydroxyethyl)-s-triazine (HTHT – CAS 4719-04-4) is registered as a MWF microbicide under the BPR, FIFRA and other nations’ pesticide regulations. However, it is also an effective sulfide scavenger used to scrub sulfide from gas generate during petroleum refining. When the product is sold for antimicrobial purposes, it has a pesticide label. When it is sold as a sulfide scavenger it has a technical chemical, it has a substantially less informative label – there is no intention of antimicrobial activity when HTHT is used as a sulfide scavenger.
For decades, I have argued the following:
- If an additive demonstrates one or more, better than average, non-biocidal functional properties – regardless of its antimicrobial properties – it need not be registered as a biocidal substance unless biocidal claims are made.
- If an additive does not demonstrate one or more, better than average, non-biocidal functional properties, and demonstrates antimicrobial performance, it should be registered as a biocidal substance.
Case study – Dicyclohexylamine
Dicyclohexylamine (DCHA, CAS 101-83-7) is a secondary amine that has been used in MWF formulations for more than two decades. As a chemical group, amines have several performance properties, the most common of which are:
- Corrosion inhibition
- Emulsion stabilization
- pH control (buffering)
However, performance in each category varies substantially among amines. When DCHA has been tested for its corrosion inhibition, emulsion stabilization, or pH control performance, it has not compared favorably relative to other amines. Figure 4 is a plot of DCHA’s antimicrobial performance in a MWF. Testing was performed per ASTM Practice E2275. As Figure 4 illustrates, in a MWF that contained DCHA at 3,000 mg kg-1 (ppm), the challenge population fell to below detection levels (BDL) and remained BDL for the duration of the eight-week study.
DCHA is an example of a chemical that has demonstrated antimicrobial performance properties, is represented as having typical amine performance properties (although with no supporting data) and is used in MWF formulations. It is a prime example of an additive that does not demonstrate one or more, better than average, non-biocidal functional properties, and demonstrates antimicrobial performance – i.e., an unregistered microbicide.
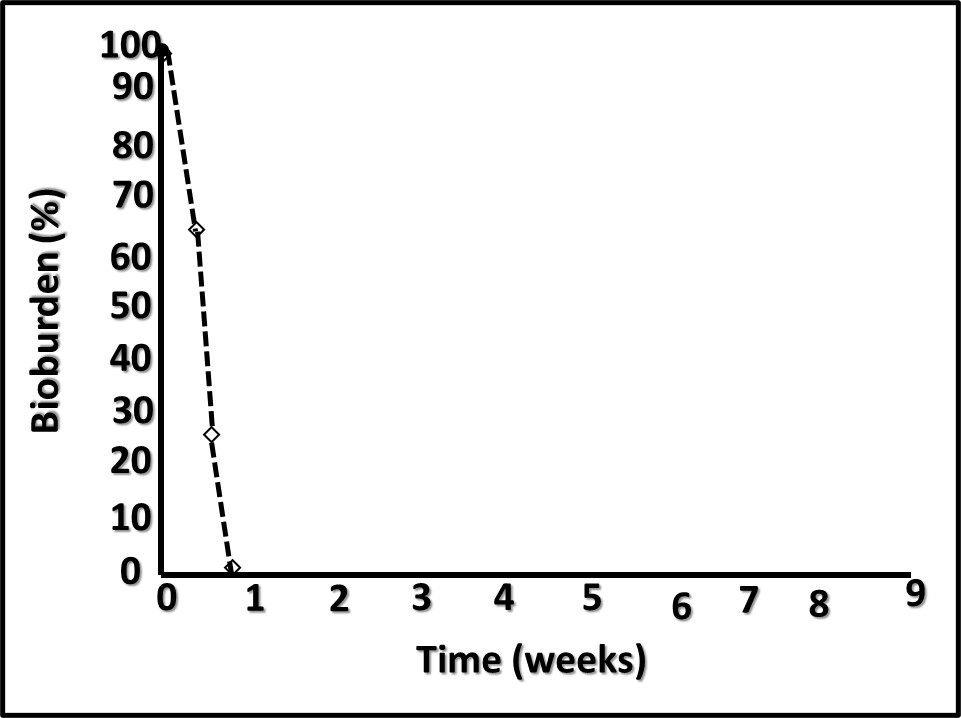
Fig 4. ASTM Practice E2275 test results – MWF formulated with DCHA at 3,000 mg kg-1.
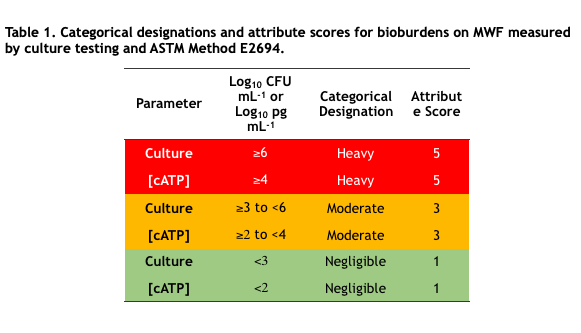
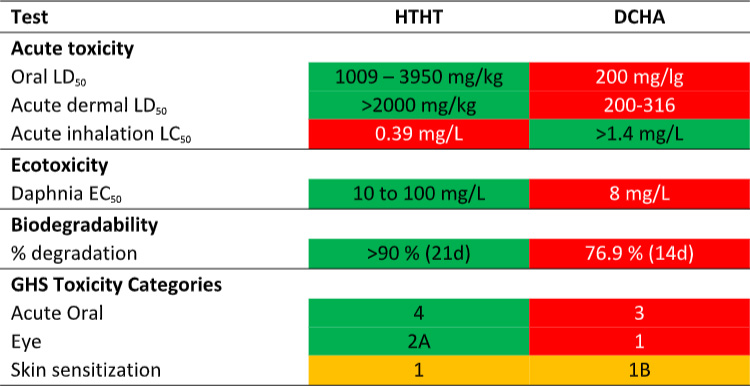
Now compare DCHA’s toxicity profile with that of HTHT. The data in Table 1 are taken from the products’ respective Safety Data Sheets (SDS). Per the SDS data, DCHA’s acute oral toxicity is >5x that of HTHT and its acute dermal toxicity is 10x that of HTHT. Moreover, DCHA’s ecotoxicity is greater than that of HTHT and its biodegradability is less than that of HTHT. Consequently, although MWF formulated with DCHA can claim to be biocide-free (they do not contain appropriately registered microbicides), they are potentially more toxic and less environmentally acceptable.
Table 1. Product SDS toxicity profile comparison – DCHA and HTHT.
Are there regulatory or liability issues?
This is an issue for regulators and lawyers. I am neither. However, there are precedents that suggest MWF compounders who use putative performance additives that do not actually demonstrate one or more, better than average, non-biocidal functional properties, but do demonstrate antimicrobial performance have exposure on both counts. There have been class-action lawsuits in which the plaintiffs have claimed adverse health effects caused by MWF exposure and in which MWF compounders have been listed as defendants. One can only speculate on the impact of formulations with unregistered microbicides on the ability of formulators to create a credible defense against adverse health complaints.
From a regulatory perspective, the issue is what claims are made. Some years ago, a food grade lubricant compounder formulated some of their products with PARABENs (para-hydroxy benzoic acid esters). Although PARABENs are commonly used as food and cosmetic preservatives, they are not registered as industrial microbicides. The compounder promoted the antimicrobial activity of their food grade lubricant. In doing so, they violated two laws. They used unregistered biocidal products as microbicides in the lubricant. They made pesticidal claims for their lubricant, although the product did not have a U.S. EPA pesticide registration. The compounder was quite fortunate in that the US EPA OPP did not press criminal charges and the fine was a fraction of what it might have been, had the US EPA’s officials applied the standard $5,000 per incident (i.e., each customer site at which product was used) per day. It has been argued that if a compounder does not claim microbial contamination resistance or other antimicrobial performance properties in their written literature, they will not come under the US EPA’s OPP scrutiny. I wonder if the risk is worth the benefit.
In terms of antimicrobial pesticides, the MWF sector is an orphan the total MWF microbicide market is estimated to be <$200 million U.S.). With the continued consolidation of biocide manufactures, and increased cost of providing all of the toxicological test data needed to support new microbicide registrations, the only new microbicides likely to be made available for use in MWF are active ingredients that have been approved in non-MWF, large volume (i.e., >$50 million opportunity for a given product) markets.
Summary
What does the term “biocide-free” mean if MWF formulated with chemistries that are more toxic than the appropriately registered antimicrobial pesticides that they replace? I suggest that all stakeholders from compounders to end-users are safer if they use additives for which complete toxicological profiles are available rather than alternatives for which only limited data are available. The increased amount of information provided on microbicide labels doesn’t make them more hazardous than other industrial chemicals. Just as a rose by any other name is would smell as sweet, a microbicidal chemical – unregistered microbicide – by any other name is just as toxic – perhaps even more so.
As always, I look forward to receiving your questions and comments at fredp@biodeterioration-control.com.
1 I originally created Figures 1, 2, and 3 for STLE’s MWF 240 Metalworking Fluid Formulation Concepts course, Module 3 Minimizing MWF Biodeterioration Risk.